

The success of chimeric antigen receptor modified T lymphocyte (CAR-T cell) therapy for pediatric and adult leukemia and lymphoma has been impressive, but far from universal. Even for the most responsive disease, pediatric pre-B-ALL, one-year remission rates are 50% or less. This brings fundamental questions as to how leukemia resists CAR-T activity front and center. CD19- and CD22-specific CAR-T escape mechanisms appear to differ. For CD19, antigen loss and RNA-splicing variants are common. For CD22, decreasing the number of target molecules on the surface of the leukemia appears to be a primary means of escape (Fry, et al., 2018). Murine models have demonstrated that bryostatin increases CD22 density and improves CAR-T activity (Ramakrishna, et al., 2019). We explored the activity of bryostatin and four FDA-approved epigenetic modifiers (panobinostat, vorinostat, 5-azacytidine, ATRA) for the ability to impact CD19 and CD22 expression in Raji, NALM6, and REH cell lines. Dose ranges tested were not toxic. For CD22: a) bryostatin, increased expression for all lines (surface molecules expressed per cell), b) 5-azacytidine, increased expression only in NALM6, and c) ATRA, increased expression only in Raji. For CD19: a) only bryostatin, and only in NALM6, increased expression. When normal B cells from peripheral blood were tested (n=3) bryostatin did not increase the number of CD19 or CD22 molecules on the cell surface.
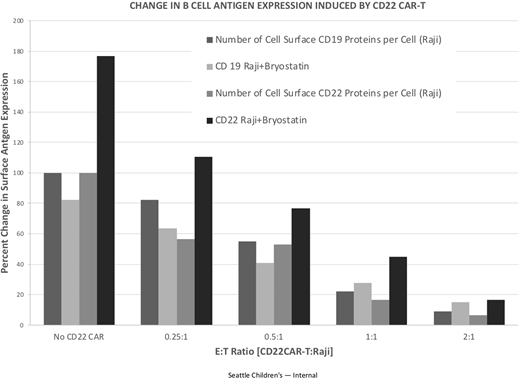
When cytolysis assays with CD22 CAR-T were carried out, bryostatin potentiated NALM6 killing, but paradoxically inhibited killing of Raji cells. To see if this effect was due to differential target molecule expression, we quantified the number of target (CD22) and non-target (CD19) molecules on the surface of each leukemia line during CTL assays. At high effector to target cell ratios, (E:T 1:1 or greater), culturing of NALM6 or Raji with CD22-CAR-T resulted in a marked reduction in both CD19 and CD22 surface expression at 24 hours, indicating a general mechanism of immune evasion. Even though CD19 and CD22 were elevated in bryostatin-treated NALM6, and CD19 was elevated in bryostatin-treated Raji, CD22 CAR-T activity still decreased target antigen expression 40% to greater than 100%. To see if this effect was due to permanent clonal selection, CAR-T were removed and purified leukemia cells re-cultured on their own. For Raji leukemia cells surviving CD22 CAR-T, the number of CD19 molecules per cell recovered within 24 hours. Full recovery was not seen for CD22, unless Raji cells had also been treated with bryostatin during the CTL assay. Our data highlights that unless bryostatin is included, CAR-T exposure induces a durable down-modulation of CD22 levels on Raji cells. We are currently defining epigenetic control of CD19 and CD22 expression upon exposure to CD22 CAR-T, bryostatin and other modifiers. These findings still do not explain why Raji cell killing is not potentiated by bryostatin, while NALM-6 is. The exposure of leukemia lines to epigenetic modifiers may also induce the expression of other molecules on the cell surface that either potentiate or inhibit CAR-T mediated killing. Preliminary data support that hypothesis that bryostatin may not only modify CD19 and CD22 expression, it may also induce both negative checkpoint molecule and positive T cell activating ligand expression on the leukemia cell surface.
Thus, CAR-T induce both general and target-antigen specific changes in leukemia, depending in part on the leukemia subtype analyzed. Bryostatin increased CD22 expression in leukemia cell lines but not in normal B cells, indicating that control of CD22 levels on the cell surface is altered by leukemic transformation. A 24-hour exposure to CD22 CAR-T can induce dramatic transient changes in CD19 and CD22 expression levels, and durable changes in CD22 expression in Raji cells that can be surmounted by the inclusion of bryostatin. The difference in Raji and NALM6 sensitization to killing may be due to the origin of these cells as Burkitt lymphoma (Raji) is more differentiated with respect to B cell lineage than pre-B ALL (NALM6). We can conclude that analysis of CAR-T target ligand number is not sufficient to predict the efficacy of CAR-T therapy. We have demonstrated that mechanisms beyond alteration in target antigen number on the cell surface, such as the induction of positive and negative T cell ligands, must also be considered in the context of the leukemia types targeted by CAR-T therapy.
Orentas:Lentigen Technology, a Miltenyi Biotec Company: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal